

This year the US has started the first measure towards a new (and long-awaited) approach to DTx reimbursement.
With the Access Prescription Digital Therapeutics Act, we may witness a change that promises to transform healthcare delivery with benefits for both digital health companies and millions of American citizens.
DTx or PDTs?
Let’s dig a little bit into this acronym challenge that may cause misunderstandings.
PDTs are Prescription Digital Therapeutics authorised by the FDA, while DTx can be considered as the larger umbrella of digital therapeutics apps and software.
When talking about DTx, we refer to a digital app that's meant to prevent, manage or treat specific medical disorders or diseases through the use of a software.
The US defines PDTs as “mobile software applications used to diagnose, treat, alleviate, and prevent diseases or other issues affecting the human body.” So, they are designed and tested much like traditional prescription drugs but rather than taking a pill, patients receive therapy through the use of software.
More than 35 PDTs have been approved by the FDA in the past five years, and some 137 products are in the pipeline.
Insurance coverage for PDTs today in the US
PDTs are subject to regulation by the Food and Drug Administration (FDA), classified according to the product’s intended use and level of risk, and subject to different degrees of oversight and requirements.
New PDTs are reviewed by the Medical Technology or Digital Formulary Committees which consider:
- FDA submission data - is a prerequisite but does not guarantee coverage
- Peer-reviewed clinical data - reliable clinical data to achieve coverage and thus justify the pricing
- HEOR data to show cost offsets or cost savings.
Today, with the Access Prescription Digital Therapeutics Act not in place, the Public Insurance Coverage is working as follows:
- Medicare does not pay for software products due to no existing appropriate benefit category for this type of solution
- Medicaid programs and care plans can consider fee-for-service product coverage via specific benefit programs on a state-by-state basis
- The Department of Defence (i.e., Veterans Affairs) is beginning to cover some digital therapeutic products with a hardware component.
The scenario on the Private Insurance side is a little different and will evolve as well in the next years. This is what it looks like today for DTx (and PDTs) solutions:
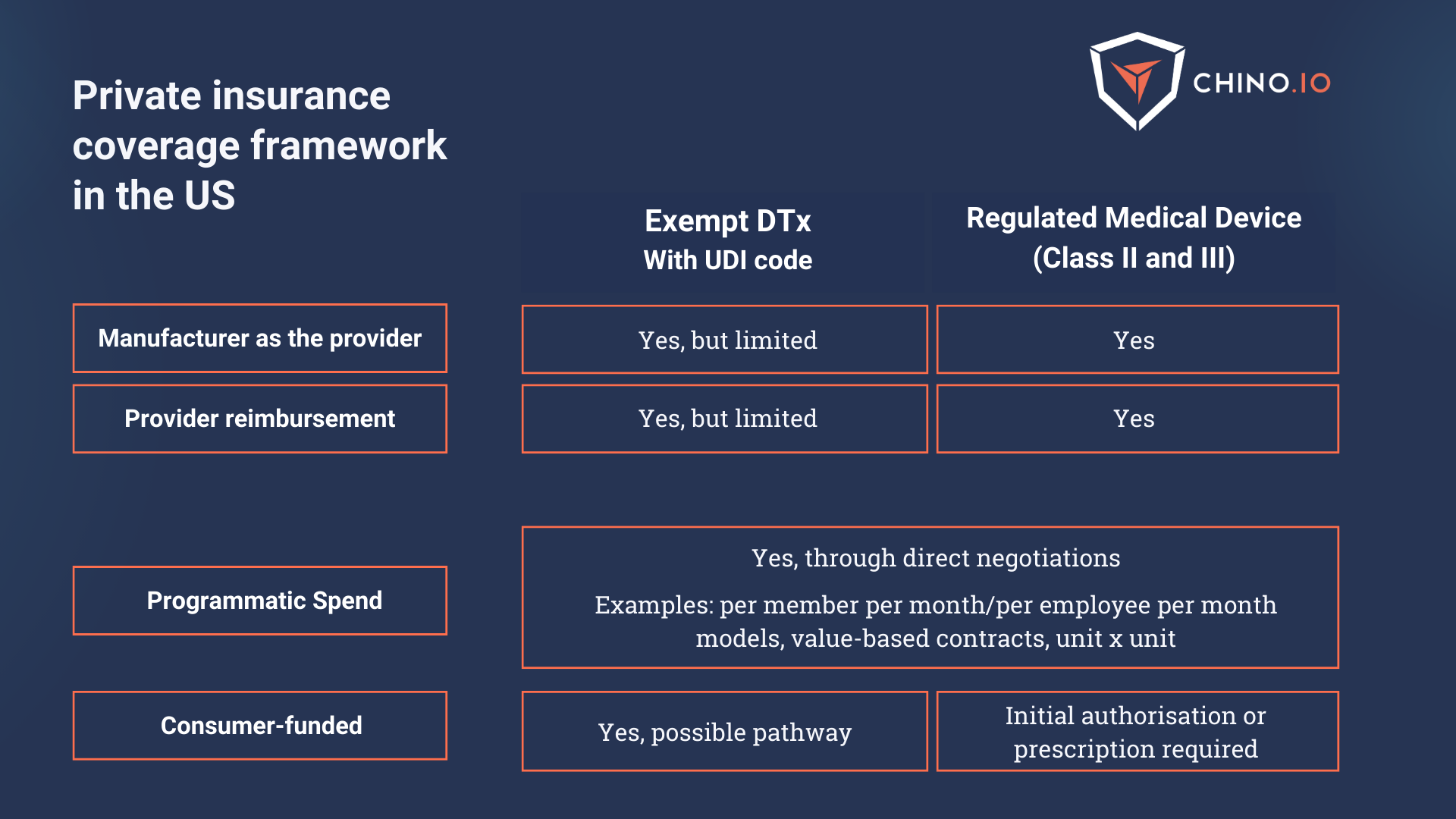
What is the Access to Prescription Digital Therapeutics Act 2023?
The Access to Prescription Digital Therapeutics Act of 2023 is a draft act that would expand coverage of PDTs, add PDTs to the list of services and products eligible for coverage under Medicare and Medicaid (health insurance programs administered by the government), and direct the Centers for Medicare and Medicaid Services (CMS) to establish payment methods and product codes for billing.
It was first introduced last year and again this year.
To be eligible for reimbursement, PDTs should meet the following criteria:
- Being cleared or approved under section 510(k), 513(f)(2), or 515 of the Federal Food, Drug, and Cosmetic Act;
- Having a cleared or approved indication for the prevention, management, or treatment of a medical disease, condition, or disorder;
- Primarily use software to achieve their intended result;
- Being exempt from section 502(f)(1) of the Federal Food, Drug, and Cosmetic Act under section 801.109 of title 21 of the Code of Federal Regulations (or any successor regulation).
According to the draft Act, PDTs manufacturers must report specified information about private payors, subject to civil penalties.
FDA approval for Software as a Medical Device
It is well known that FDA approval and audits are not easy. Preparation to submit and maintain the FDA approval requires effort and expertise. So get ready (especially if you have an innovative product with no prior examples).

PDTs - and all Software as a Medical Device (SaMD) products - are evaluated for their perceived potential risk to patients and assigned to one of three classes. As you will see, all classes of devices are subject to General Controls, as are the baseline requirements.
- Class I - low risk: here, we find devices showing that general controls of the act are sufficient to ensure safety and effectiveness. Most Class I devices are exempt from Premarket Notification 510(k).
- Class II - moderate risk: requires general regulatory controls and often special regulatory controls, such as a requirement for clinical data specific to a product in order to provide reasonable assurance of safety and effectiveness or to demonstrate substantial equivalence to a predicate device. Most Class II devices require Premarket Notification 510(k).
- Class III - high risk: require general controls and Premarket Approval (PMA) - The PMA is the most stringent type of device marketing application required by the FDA.
Class II devices generally include special controls. For example, the FDA might specify requirements around labeling or clinical data to satisfy questions of safety and effectiveness.
While the majority of approved PDTs in the US are classified as class I or class II medical devices according to their indications, other solutions may fall into different risk classes depending on their treatment areas.
Another characteristic of SaMD is the Code of Federal Regulations (CFR). It lists a variety of regulations regarding computerised therapies that are unique to the diseases that a particular SaMD product is designed to treat. For this reason, the regulations are intended as fit for purpose. For example:
- Software developed for psychiatric disorders follows the requirements for Computerised Behavioral Therapy device for psychiatric disorders (21 CFR 882.5801).
- Software for gastrointestinal conditions follows the requirements for Computerised Behavioral Therapy device for treating symptoms of gastrointestinal conditions (21 CFR 876.5960).
So, what are the main steps to get the approval?
If you want to market your PDTs product, first of all, you will need to carry out one or more clinical studies (e.g., randomised controlled trials).
Once it has been evaluated (with a positive outcome, of course 😉), you can submit the data and formal requests for authorisation via one of two FDA pathways (each with regulatory and evidence-based requirements):
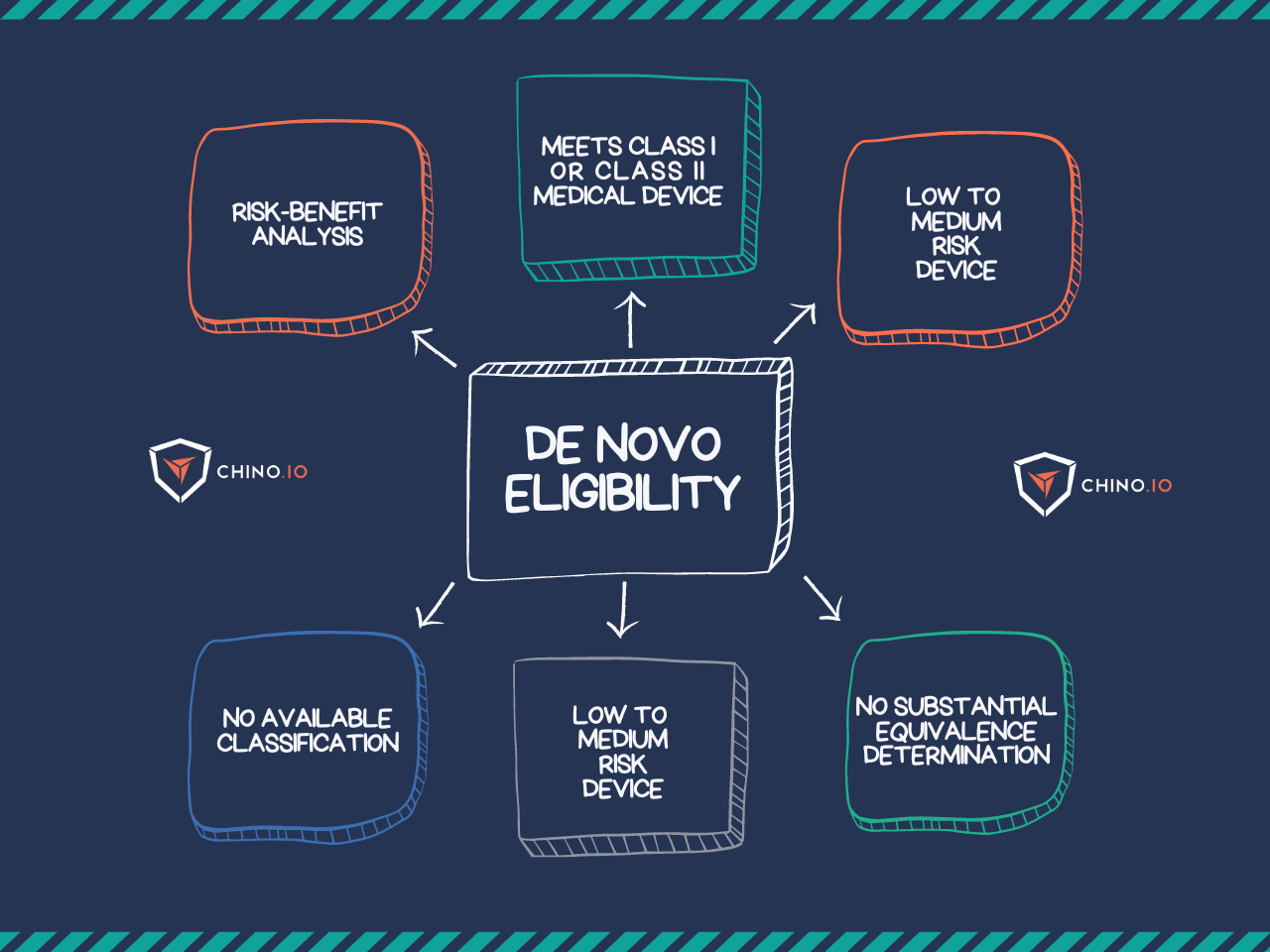
• De novo pathway: it requires clinical data demonstrating the safety and effectiveness of the device. Devices authorised via this pathway can then serve as “predicates” for other devices.
• 510(k) clearance pathway: requires the submission of clinical data demonstrating substantial equivalence in terms of safety and effectiveness to a predicate product authorised either via the de novo or another 510(k) pathway.
Unlike pharmaceuticals, SaMD products can be frequently updated following FDA authorisation, as products relying on AI as a component of treatment may “learn” or change how their algorithms perform over time.
Work is currently underway to create a dedicated FDA regulatory framework for SaMD products (such as PDTs) that reflects the unique attributes of these devices.
What can you do to be ready to access the US market?
This is an evolving and fragmented regulatory framework that may seem (and actually is) challenging for companies and startups.
- Check if HIPAA compliance applies: to comply with HIPAA (and the GDPR), make sure to implement appropriate organisational (legal/administrative) and technical security requirements. And ensure that your service providers (also called Data-Processors in the EU or Business Associates in the US) comply with the GDPR or HIPAA as well. HIPAA requirements are also similar to the GDPR ones, so if you have already done your homework, this will be easier for you.
- Work on quality management system standards: the FDA requires companies to go through a pre-certification program to be recognized by regulators. Regulated as Software as a Medical Device (also known as SaMD), approval is subject to specific quality management system standards for software development and clinical evaluation, among other requirements.
- Demonstrate your ability to keep data secure: as you will be asked to provide evidence of aspects such as a robust quality management system, be ready to show your ability to keep data secure. This helps ensure patients' safety while leaving you free to improve your products. Thus, we suggest you implement technical security measures. If you did GDPR before, you probably have it covered already. Getting a certification as the ISO 27001 for example, can also help you to increase the trust towards your product and your company.
Developers' compliance with FDA regulations and best practices is critical to maintain trust and transparency and reduce the risk of harm. Organisations and stakeholders, including payers, provider organisations, clinicians, and developers, have a responsibility to their patients to use products that are safe and effective. Maintaining trust and transparency is critical for patients and public health.
If you decide to enter the US (and any other EU country) with your DTx product, be sure to have a solid compliance baseline set. It will help you to reduce time and costs when marketing your solution.
Don't forget to read our article about the reimbursement path for DTx in Europe!
Chino.io, your trusted compliance partner
Working with experts can reduce time-to-market and technical debt and ensure a clear roadmap you can showcase to partners and investors (see our latest case study).
At Chino.io, we have been combining our technological and legal expertise to help hundreds of companies like yours navigate through EU and US regulatory frameworks enabling successful launches and reimbursement approvals.
We offer tailored solutions to support you in meeting the GDPR, HIPAA, DVG, or DTAC requirements mandated for listing your product as DTx/PDTs.
Want to know how we can help you? Reach out to us and learn more.

Subscribe to the Chino.io newsletter to get monthly updates on compliance, data privacy, and security.



